Master Bioinfo Projets et Stages
Désolé, cette activité n’est pas visible actuellement
Aperçu des sections
-
-
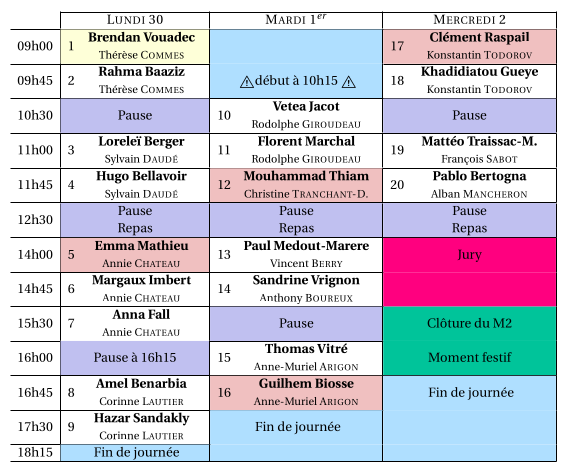
-
Annonces Forum
-
Offres de stage 2024-25 Dossier
-
Offres de stage spéciales M1 2024-25 Dossier
-
Affectation tuteurs et tutrices M1 et M2 AS Fichier
-
Affectation tuteurs et tutrices M2 Fichier
-
-
-
-
-
-
